
Prof. dr. Jeroen Pasterkamp
PI

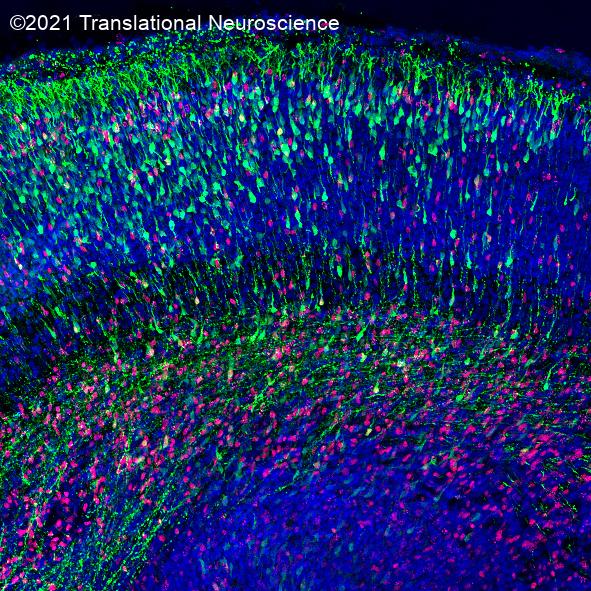
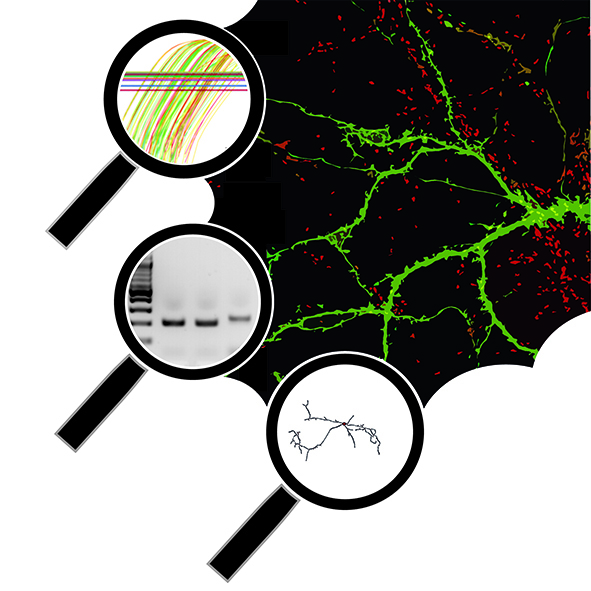
I am a molecular neurobiologist trying to dissect the mechanisms that underlie neural circuit development and various neurological and neurodegenerative disorders using a multidisciplinary research approach ranging from in vitro human stem cell modelling and mouse genetics to advanced 3D microscopy.